









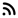


 19:34 / 31.07.2024
19:34 / 31.07.2024 3541
3541 Фенилкетонурия представлява автозомно-рецесивно генетично нарушение на метаболизма на фенилаланин, което възниква в резултат на дефицит на фенилаланинхидроксилаза (PAH). Повечето форми на фенилкетонурия (PKU) и по-лекият ѝ вариант хиперфенилаланинемия (HPA) се причиняват от мутации в гена на PAH на хромозома 12q23.2.
Заболяването се проявява около 2 месец след раждането на бебето с неспокойствие, повръщане, обрив. Децата най-често са добре охранени, което заблуждава родителите. В повечето случаи (до 90%) децата имат руса коса и сини очи, нежна и чувствителна кожа. Мускулният тонус се променя, наблюдават се повишени сухожилни рефлекси, патологични рефлекси от групата на Бабински.
Засегнатите от фенилкетонурия имат специфична миризма на урината, наподобяваща миризмата на мишка, микроцефалия, забавено развитие на говора и генерализирани гърчове. Поведението също е засегнато под формата на агресия, автоагресия, аутизъм. Гърчовете се появяват след 6-месечна възраст и протичат с поклащане на главата, кимане, свиване на тялото напред, пише Puls.bg.
Нелекуваната фенилкетонурия се свързва с нарушения, които включват забавяне на растежа, лоша пигментация на кожата, гърчове, забавяне на развитието и тежко интелектуално увреждане. В резултат на недостиг или на намалена активност на ензима фенилаланинхидроксилаза (PAH), фенилаланинът не може да се превърне в тирозин. Това води до натрупване на фенилаланин и последващото му разграждане до други продукти по алтернативни пътища (фенилпируват, фенилацетат, фениллактат, които се отделят чрез урината). От друга страна, нивото на тирозин е ниско, което предизвиква намалена синтеза на невротрансмитери.
Как се поставя диагноза за фенилкетонурия?
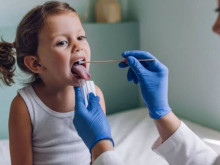
Диагнозата се базира на увеличени серумни концентрации на фенилаланин. Нормалната концентрация на ензима е от 1-2.5 mg/dl. Взима се малко количество кръв (обикновено от петата на новороденото) за изследване. Нивата на фенилаланин в кръвта започват да се повишават до 24 часа след захранване на бебето, затова тестът за изследване за фенилкетонурия е необходимо да бъде извършен до 2-3 дни след раждането. На 7–10 ден се прави контролно изследване.
Какво е лечението за фенилкетонурия?
Лечението на фенилкетонурията се базира единствено на следването на диета, бедна на фенилаланин. Забранява се приема на храни, в които се съдържа тази аминокиселина - месо, мляко, млечни продукти, брашно, ядки, шоколад. Необходимо е да се избягват напитки, съдържащи изкуствени подсладители, защото някои от тях съдържат фенилаланин.
Какво е фенилкетонурия?
©






Още от категорията

















| | ||||||
|---|---|---|---|---|---|---|
| пон | вто | сря | чтв | пет | съб | нед |
| 1 | 2 | 3 | ||||
| 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 11 | 12 | 13 | 14 | 15 | 16 | 17 |
| 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| 25 | 26 | 27 | 28 | 29 | 30 | |

Директорът на "Асоциация Анимус": Случаите с домашно насилие став...
21:27 / 25.11.2024

Елена Поптодорова за изборите в Румъния: Появи се черен лебед! То...
20:58 / 25.11.2024

Проф. Боян Дуранкев: Ако България влезе в Шенген, ще ни имат дове...
19:48 / 25.11.2024

Психолог за случая в столичния мол: Тези случаи говорят за това, ...
19:49 / 25.11.2024

Андрей Райчев: В парламента има жалки усилия на оцеляващи хора да...
19:26 / 25.11.2024

Външният ни министър се срещна със специалния представител на Гер...
19:23 / 25.11.2024
Актуални теми

